September 2018
By Alberto Ortín, Pilar del Hierro. Polymer Char, Valencia, Spain.
New research leads to the development of an automated and versatile technique for intrinsic viscosity (or solution viscosity) determination of a wide range of polymers in different solvents, from ambient temperature up to 200ºC. The technique uses a two-capillary serial viscometer that collects relative viscosity data from which intrinsic viscosity is derived. The serial capillaries design is self-cleaning and self-calibrating, ensuring reliable and precise data. This new process becomes a practical and convenient alternative to manual or semi-automated methods.
Introduction
The determination of the solution viscosity of polymeric materials is very important to the industry, both to research and manufacturing, since it can be used to estimate the molar mass, providing important information relating to the physical properties of polymers. The relative viscosity of a dilute polymer solution to that of the pure solvent itself is measured and, from it, the Intrinsic Viscosity (IV or [η]) of the polymer is calculated.
Due to the popularity of dilute solution viscosity measurements, and the availability of those methods in many manufacturing laboratories, the intrinsic viscosity of polymers has been traditionally used to specify and to control the production grades. It must be noted however, that intrinsic viscosity is not a property of the polymer itself, as molar mass is, but rather a property of the polymer solution, influenced by the solvent and temperature.
The recently developed technique is an automated process for viscosity measurements of polymeric materials in solution. It is compatible with typically used organic solvents such as decalin, tetralin, tri-chlorobenzene (TCB) and ortho-dichlorobenzene among many others. Dissolution temperature and analysis temperature can be set independently, from ambient to 200ºC, so that a wide range of polymers, even the most crystalline ones, can be analyzed with convenience and safety with this alternative approach.
Intrinsic Viscosity Determination
The proposed new method performs the polymer intrinsic viscosity measurement by means of a two-capillary relative viscosity detector, concept that was developed and patented by Yau in the 80s (US 4,793,174), as a robust method in contrast to temperature, pressure or solvent flow-rate variations.
Capillary viscometers rely on the principle that under a forced flow (Q), the pressure drop (ΔP) due to a fluid traveling along a capillary tubing of length L and radius r, is proportional to the absolute viscosity of the flowing fluid (η), according to Poiseuille’s law:
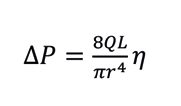
Equation 1. Poiseuille’s Law
Absolute viscosity of fluids is important to many industries and can be measured using different types viscometers. However, in polymeric materials, the interest is on the relative viscosity of a dilute polymer solution compared to that of the pure solvent, given that from it, the intrinsic viscosity, related to molar mass of the polymeric material, can be derived.
First, the relative viscosity (ηrel) is defined as:
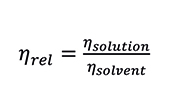
Equation 2. Relative Viscosity
This is a dimensionless quantity which represents to what extent the added polymeric material increases the viscosity of the solvent. The relative viscosity of the solution is proportional to the amount or concentration (C) of polymer that it contains, while the intrinsic viscosity is independent of concentration. The specific viscosity (ηsp) of the solution and the polymer intrinsic viscosity are calculated according to the Equations:
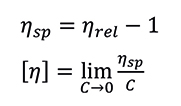
Equation 3 (above): Specific Viscosity. Equation 4 (below): Intrinsic Viscosity
The intrinsic viscosity has units of inverse density (dL/g for instance). It is defined at the limit of infinite dilution (zero concentration), and sometimes calculated by extrapolation of data at different concentration levels. A more practical and efficient approach is based on a single-point relative viscosity measurement, taken at a defined concentration low enough to eliminate the need for extrapolation, or using a model equation to estimate the extrapolated value. Among several models and equations, the Solomon-Ciutà Equation, which does not require additional parameters, can be used:
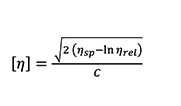
Equation 5. Solomon-Ciutà Equation
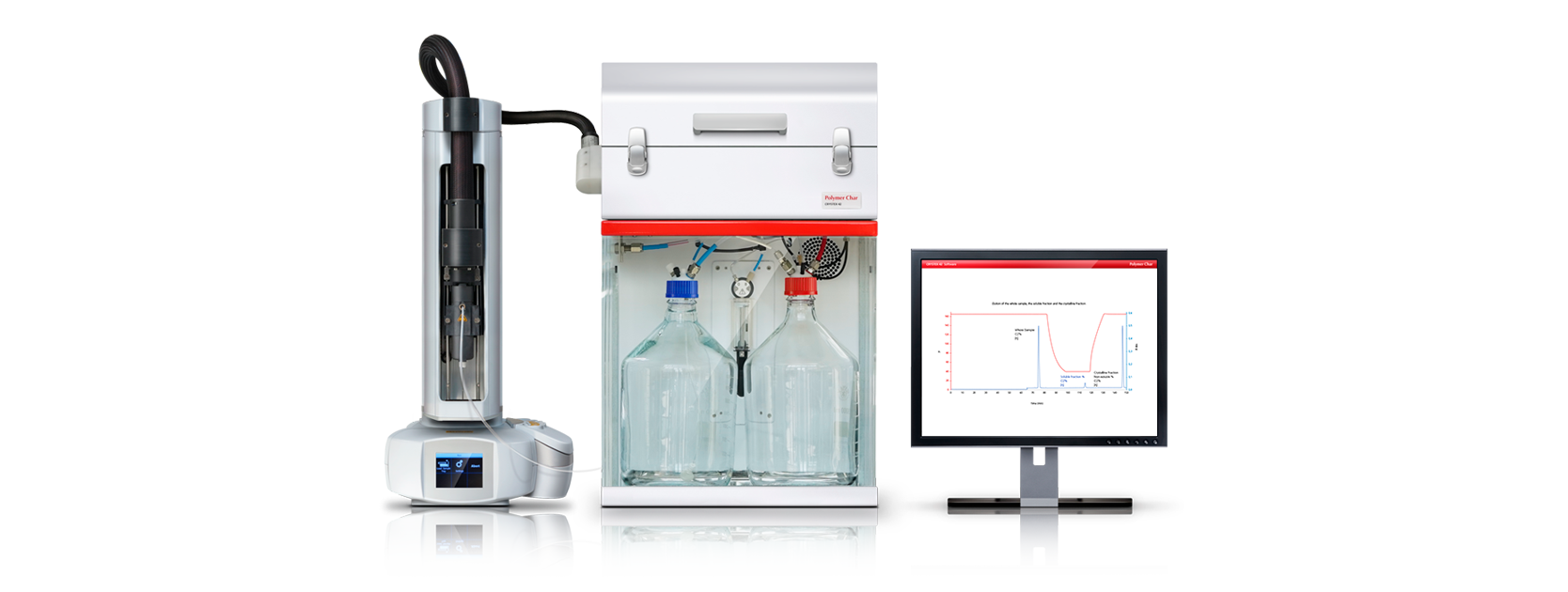
A serial viscometer design has been implemented for the new method: two pieces of capillary tubing are connected in series in such a way that the first one experiments the flow of pure solvent while the second one simultaneously receives the flow of the dilute polymer solution. The pressure drop across each capillary is measured continuously as a function of time by high sensitivity differential pressure transducers.
According to Equations (1) and (2), the relative viscosity is proportional to the ratio of differential pressures, being independent of the flow rate. An instrumental constant is easily measured by flowing pure solvent through the two capillaries. The constant is automatically measured with every injection thus, compensating for small, long-term variations in capillaries (self-calibrating). It also accounts for differences in tubing dimensions, so in this design, it is not required that the capillaries stay accurately matched.
The relative viscosity is calculated as a function of time as the injected solution goes through the system, and any influence of flow-rate variations (such as high frequency pump pulsations) or thermal effects are cancelled out directly by the reference capillary, resulting in very high sensitivity and long-term stability.
An automated technique for Intrinsic Viscosity Analysis
The proposed method constitutes a precise and convenient approach to intrinsic viscosity measurement, due to the automation of all the analytical procedures, from filling of vials to the reporting of results. Samples are put in solid form into 20 mL vials and brought to an autosampler tray with capacity of 42 vials. The operator enters the samples identification data, selects the analytical method and starts the analysis that proceeds unattended until all the vials defined in the instrument run queue are analyzed. Under software control, solvent is added to the vials, controlling the dissolution time per vial, injecting each solution and rinsing of the capillary lines. The solution travels safely throughout all the system without any risk of precipitation because there are no cold spots. A new run can be immediately started after finishing the previous one, achieving a throughput of 40 samples a day in standard operation conditions.
In order to maintain the polymeric sample integrity along the dissolution and measurement processes, Sample Care protocols have been implemented as part of the method. Those include the ability to purge the vial atmosphere with an inert gas (Nitrogen) before dissolution starts, and controlling accurately the time spent at high temperature by keeping every vial inside of the oven only for the programmed dissolution time. The vials remain in an external tray at room temperature until the scheduling software requests their transfer to the dissolution oven. Efficient heat transfer to the vial, together with preheating of the solvent prior to delivery to the vial, help in shortening the time required for dissolution. Oxidative and thermal degradation is thus minimized, ensuring that accurate intrinsic viscosity is measured even for the most challenging ultra-high molar mass materials, or oxidation-prone polymers, such as polypropylene.
Results and Applications
Different polymers in various solvents have been analyzed in this system, including PAN (polyacrylonitrile) in DMF (N,N-dimethylformamide), PET in phenol:o-DCB, PLA in TCB as well as polyolefins in TCB and o-DCB.
Even if the procedure or conditions in this new system were different, the intrinsic viscosity results obtained were in good agreement with reference methods (ISO 1628-3:2010 f.i. and ASTM D1601) in all cases, as shown in Figures 1 and 2 for two of the systems considered.
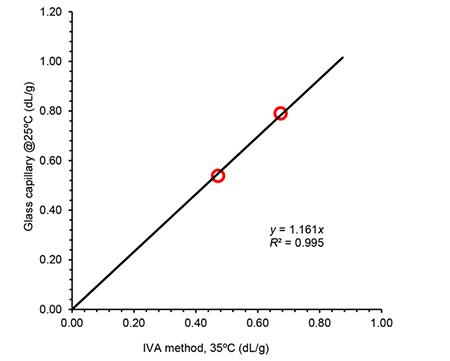
Figure 1. Correlation of intrinsic viscosity (IV) values obtained using the IVA method compared to IV determined using a traditional Ubbelohde type glass capillary setup for a set of PET samples. In both cases a mixture of phenol:o-DCB (2:3, m:v) was used while temperatures were 35ºC and 25ºC respectively.
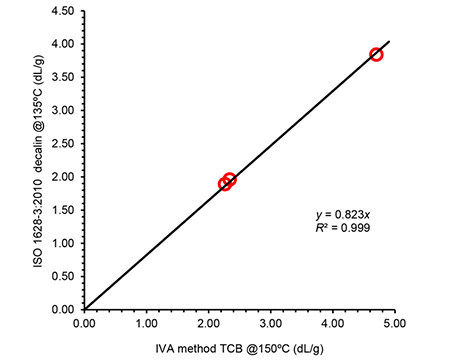
Figure 2. Correlation of intrinsic viscosity (IV) values obtained using the IVA method compared to IV determined according to standard method ISO 1628-3:2010 for a set of polypropylene samples. IVA was run in tri-chlorobenzene at 150ºC, using solutions prepared at 1 mg/mL; the ISO method was performed in decalin at 135ºC with solutions at 1-1.5 mg/mL. The linear correlation is excellent as demonstrated by the high R2 value.
A factor to keep in mind when comparing different methods to evaluate the intrinsic viscosity in polymeric materials is the effect of shear rate on the observed viscosity. Methods based on glass capillaries, such as the Ubbelohde type viscometer, are driven by gravity and operate under low shear condition, while forced flow, used in this new method, may work at higher shear rates. Typically when shear rate increases the observed viscosity decreases, so keeping constant shear rate may be advantageous in order to get consistent results.
A study was conducted for a set of PE materials spanning a very wide range in IV, in order to compare the observed viscosity at two different flow rates. Results depicted in Figure 3 show that an excellent correlation was found, and therefore a high flow-rate determination can be performed in order to reduce the analysis time, and still obtain data comparable to lower shear conditions. The analysis time per sample was reduced from half an hour to 4 minutes with the increased flow rate.
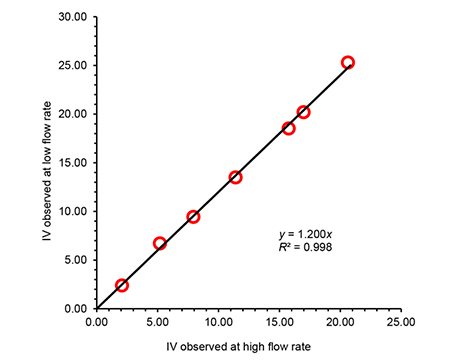
Figure 3. Observed intrinsic viscosity for a series or polyethylene materials, in trichlorobenzene at 150ºC, using high and low flow-rate through the two-capillary viscometer.
Application example: Intrinsic Viscosity of UHMWPE
Maybe one of the most challenging polymers for analysis is ultra-high molar mass polyethylene (UHMWPE). Those are high crystallinity materials with extremely high values of IV, only soluble at elevated temperatures in organic solvents. Special care must be taken in sample preparation to prevent degradation, which would reduce the apparent viscosity, and also in the analysis, given the high viscosity of the solutions.
Dissolution temperature and time must be chosen so that the polymer dissolves completely but also to give the very long molecular chains enough time to disentangle completely and obtain reliable viscosity values. In Figure 4, data collected after 90 minutes or 180 minutes of dissolution show that longer time is required to achieve that full disentanglement condition; otherwise, the intrinsic viscosity is underestimated. When the polymer stays at elevated temperature for such a long time, a nitrogen atmosphere is also required to prevent oxidative degradation which also decreases the observed viscosity as seen in the same Figure 4.

Figure 4. Observed viscosity for an UHMWPE sample with different dissolution time, with and without nitrogen purge of the dissolution vial.
An Infrared (IR) detector can be used for accurate quantification of the injected mass, which otherwise needs to be weighed using an analytical balance. The IR detector is suitable for detection of polymers with aliphatic CH groups, dissolved and analyzed in solvents that are IR transparent, such as the important class of polyolefins (polyethylene, polypropylene and copolymers), in TCB or o-DCB
In order to demonstrate the advantage in precision when applying online IR quantification compared to the offline balance, three different high molar mass PE materials were analyzed at a low concentration level of 0.25 or 0.15 mg/mL in TCB. Dissolution time was 1 hour with gentle shaking, at 140ºC under nitrogen atmosphere, in order to minimize thermal and oxidative degradation. Results are presented in Table 1, together with standard deviation, when using the IR detector for quantifying the injected mass, and when using the nominal weight given by the analytical balance, in the IV calculation. An improvement in the precision is clearly seen when the actual mass measured by the IR detector is considered, given it eliminates any errors associated with handling small amounts of polymer by the operator, but also due to the possible presence of non-soluble particles.